June 16, 2014
Actelion Ltd today announced the top-line results of the pivotal Phase III GRIPHON study in 1,156 patients with pulmonary arterial hypertension (PAH) with selexipag, the first selective oral prostacyclin IP receptor agonist. Initial analysis shows that the event-driven outcome study has met its primary efficacy endpoint with high statistical significance.
June 16, 2014
Actelion Ltd today announced the top-line results of the pivotal Phase III GRIPHON study in 1,156 patients with pulmonary arterial hypertension (PAH) with selexipag, the first selective oral prostacyclin IP receptor agonist. Initial analysis shows that the event-driven outcome study has met its primary efficacy endpoint with high statistical significance.
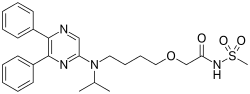
Selexipag
N-[2-[4-[N-(5,6-Diphenylpyrazin-2-yl)-N-isopropylamino]butoxy]acetyl]methanesulfonamide
2-[4-[N-(5,6-Diphenylpyrazin-2-yl)-N-isopropylamino]butoxy]-N-(methylsulfonyl)acetamide
phase 3 pulmonary hypertention
Selexipag (ACT-293987, NS-304) is a drug currently in development by Actelion as a treatment of pulmonary arterial hypertension. Selexipag and its active metabolite, ACT-333679, are agonists at the PGI2 prostaglandin receptor, which leads to vasodilation in the pulmonary circulation
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of
5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors.
In April 2008, Actelion and Nippon Shinyaku signed a licensing agreement, under which Actelion will be responsible for the global development and commercialization of selexipag outside Japan, and the two companies will co-develop and co-commercialize the drug in Japan.
http://www1.actelion.com/sites/en/scientists/development-pipeline/phase-3/selexipag.page
ABOUT THE ACTELION / NIPPON SHINYAKU ALLIANCE
Actelion and Nippon Shinyaku entered into an exclusive worldwide alliance in April 2008 to collaborate on selexipag, a first-in-class orally-available, selective IP receptor agonist for patients suffering from pulmonary arterial hypertension (PAH). This compound was originally discovered and synthesized by Nippon Shinyaku. Phase II evaluation has been completed, and a Phase III program in PAH patients has been initiated. Actelion is responsible for global development and commercialization of selexipag outside Japan, while the two companies will co-develop and co-commercialize in Japan. Nippon Shinyaku will receive milestone payments based on development stage and sales milestones as well as royalties on any sales of selexipag.
| Selexipag | |
|---|---|
|
|
| Identifiers | |
| CAS number | 475086-01-2  |
| PubChem | 9913767 |
| ChemSpider | 8089417 |
| UNII | 5EXC0E384L |
| KEGG | D09994 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C26H32N4O4S |
| Molar mass | 496.6 g·mol−1 |
NS-304 (ACT-293987), an orally available long acting non-prostanoid prostaglandin I2 (PGI-2) receptor agonist, is in phase III clinical trials at Actelion for the oral treatment of pulmonary hypertension. Nippon Shinyaku is conducting phase III clinical trials with NS-304 for this indication in Europe. In Japan, phase II clinical trials are ongoing for the treatment of pulmonary hypertension and chronic thromboembolic pulmonary hypertension.
Originally discovered and synthesized by Nippon Shinyaku, NS-304 stimulates PGI-2 receptors in blood vessels and exerts vasodilating effects.
In 2008, the compound was licensed to Actelion by Nippon Shinyaku on a worldwide basis with the exception of Japan for the oral treatment of pulmonary arterial hypertension (PAH). According to the final licensing agreement, Actelion will be responsible for global development and commercialization of NS-304 outside Japan, while the two companies will codevelop and co-commercialize the product candidate in Japan. In 2005, orphan drug designation was assigned in the E.U. by Nippon Shinyaku for the treatment of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension.
…………………….
US2012/101276
http://www.google.st/patents/US20120101276?hl=pt-PT&cl=en
The present invention relates to a crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide (hereinafter referred to as “compound A”).

BACKGROUND OF THE INVENTION
Compound A has an excellent PGI2 agonistic effect and shows a platelet aggregation inhibitory effect, a vasodilative effect, a bronchodilative effect, a lipid deposition inhibitory effect, a leukocyte activation inhibitory effect, etc. (see, for example, in WO 2002/088084 (“WO ‘084”)).
Specifically, compound A is useful as preventive or therapeutic agents for transient ischemic attack (TIA), diabetic neuropathy, diabetic gangrene, peripheral circulatory disturbance (e.g., chronic arterial occlusion, intermittent claudication, peripheral embolism, vibration syndrome, Raynaud’s disease), connective tissue disease (e.g., systemic lupus erythematosus, scleroderma, mixed connective tissue disease, vasculitic syndrome), reocclusion/restenosis after percutaneous transluminal coronary angioplasty (PTCA), arteriosclerosis, thrombosis (e.g., acute-phase cerebral thrombosis, pulmonary embolism), hypertension, pulmonary hypertension, ischemic disorder (e.g., cerebral infarction, myocardial infarction), angina (e.g., stable angina, unstable angina), glomerulonephritis, diabetic nephropathy, chronic renal failure, allergy, bronchial asthma, ulcer, pressure ulcer (bedsore), restenosis after coronary intervention such as atherectomy and stent implantation, thrombocytopenia by dialysis, the diseases in which fibrosis of organs or tissues is involved [e.g., Renal diseases (e.g., tuburointerstitial nephritis), respiratory diseases (e.g., interstitial pneumonia (pulmonary fibrosis), chronic obstructive pulmonary disease), digestive diseases (e.g., hepatocirrhosis, viral hepatitis, chronic pancreatitis and scirrhous stomachic cancer), cardiovascular diseases (e.g, myocardial fibrosis), bone and articular diseases (e.g, bone marrow fibrosis and rheumatoid arthritis), skin diseases (e.g, cicatrix after operation, scalded cicatrix, keloid, and hypertrophic cicatrix), obstetric diseases (e.g., hysteromyoma), urinary diseases (e.g., prostatic hypertrophy), other diseases (e.g., Alzheimer's disease, sclerosing peritonitis; type I diabetes and organ adhesion after operation)], erectile dysfunction (e.g., diabetic erectile dysfunction, psychogenic erectile dysfunction, psychotic erectile dysfunction, erectile dysfunction associated with chronic renal failure, erectile dysfunction after intrapelvic operation for removing prostata, and vascular erectile dysfunction associated with aging and arteriosclerosis), inflammatory bowel disease (e.g., ulcerative colitis, Crohn’s disease, intestinal tuberculosis, ischemic colitis and intestinal ulcer associated with Behcet disease), gastritis, gastric ulcer, ischemic ophthalmopathy (e.g., retinal artery occlusion, retinal vein occlusion, ischemic optic neuropathy), sudden hearing loss, avascular necrosis of bone, intestinal damage caused by administration of a non-steroidal anti-inflammatory agent (e.g., diclofenac, meloxicam, oxaprozin, nabumetone, indomethacin, ibuprofen, ketoprofen, naproxen, celecoxib) (there is no particular limitation for the intestinal damage so far as it is damage appearing in duodenum, small intestine and large intestine and examples thereof include mucosal damage such as erosion and ulcer generated in duodenum, small intestine and large intestine), and symptoms associated with lumbar spinal canal stenosis (e.g., paralysis, dullness in sensory perception, pain, numbness, lowering in walking ability, etc. associated with cervical spinal canal stenosis, thoracic spinal canal stenosis, lumbar spinal canal stenosis, diffuse spinal canal stenosis or sacral stenosis) etc. (see, for example, in WO ‘084, WO 2009/157396, WO 2009/107736, WO 2009/154246, WO 2009/157397, and WO 2009/157398).
In addition, compound A is useful as an accelerating agent for angiogenic therapy such as gene therapy or autologous bone marrow transplantation, an accelerating agent for angiogenesis in restoration of peripheral artery or angiogenic therapy, etc. (see, for example, in WO ‘084).
Production of Compound A
Compound A can be produced, for example, according to the method described in WO ‘084, and, it can also be produced according to the production method mentioned below.

Step 1:
6-Iodo-2,3-diphenylpyrazine can be produced from 6-chloro-2,3-diphenylpyrazine by reacting it with sodium iodide. The reaction is carried out in the presence of an acid in an organic solvent (e.g., ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or their mixed solvent). The acid to be used is, for example, acetic acid, sulfuric acid, or their mixed acid. The amount of sodium iodide to be used is generally within a range of from 1 to 10 molar ratio relative to 6-chloro-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the acid to be used, but may be generally within a range of from 60° C. to 90° C. The reaction time varies depending on the kinds of the solvent and the acid to be used and on the reaction temperature, but may be generally within a range of from 9 hours to 15 hours.
Step 2:
5,6-Diphenyl-2-[(4-hydroxybutyl(isopropyl)amino]pyrazine can be produced from 6-iodo-2,3-diphenylpyrazine by reacting it with 4-hydroxybutyl(isopropyl)amine. The reaction is carried out in the presence of a base in an organic solvent (e.g., sulfolane, N-methylpyrrolidone, N,N-dimethylimidazolidinone, dimethyl sulfoxide or their mixed solvent). The base to be used is, for example, sodium hydrogencarbonate, potassium hydrogencarbonate, potassium carbonate, sodium carbonate or their mixed base. The amount of 4-hydroxybutyl(isopropyl)amine to be used may be generally within a range of from 1.5 to 5.0 molar ratio relative to 6-iodo-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from 170° C. to 200° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 5 hours to 9 hours.
Step 3:
Compound A can be produced from 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine by reacting it with N-(2-chloroacetyl)methanesulfonamide. The reaction is carried out in the presence of a base in a solvent (N-methylpyrrolidone, 2-methyl-2-propanol or their mixed solvent). The base to be used is, for example, potassium t-butoxide, sodium t-butoxide or their mixed base. The amount of N-(2-chloroacetyl)methanesulfonamide to be used may be generally within a range of from 2 to 4 molar ratio relative to 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from −20° C. to 20° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 0.5 hours to 2 hours.
The compounds to be used as the starting materials in the above-mentioned production method for compound A are known compounds, or can be produced by known methods.
…………………………………
WO 2002088084
and
http://www.google.fm/patents/WO2009157398A1?cl=en
………………………
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 21 p. 6692 – 6704
compd 31
……………………
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 24 p. 7720 – 7725
 2a isthe drug
2a isthe drug
N-Acylsulfonamide and N-acylsulfonylurea derivatives of the carboxylic acid prostacyclin receptor agonist 1 were synthesized and their potential as prodrug forms of the carboxylic acid was evaluated in vitro and in vivo. These compounds were converted to the active compound 1 by hepatic microsomes from rats, dogs, monkeys, and humans, and some of the compounds were shown to yield sustained plasma concentrations of 1 when they were orally administered to monkeys. These types of analogues, including NS-304 (2a), are potentially useful prodrugs of 1.
http://www.sciencedirect.com/science/article/pii/S0968089607007614
References
- Kuwano et al. NS-304, an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007;322:1181-1188.
- Kuwano et al. A long-acting and highly selective prostacyclin receptor agonist prodrug, NS-304, ameliorates rat pulmonary hypertension with unique relaxant responses of its active form MRE-269 on rat pulmonary artery. J Pharmacol Exp Ther 2008;326:691-699.
- Simonneau G, Lang I, Torbicki A, Hoeper MM, Delcroix M, Karlocai K, Galie N. Selexipag, an oral, selective IP receptor agonist for the treatment of pulmonary arterial hypertension Eur Respir J 2012; 40: 874-880
- Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010;104:9-21.
- Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
ABOUT SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
Filed under: 0rphan drug status, Phase3 drugs, Uncategorized Tagged: ACT-293987, NS-304, PHASE 3, Selexipag







