

Female Libido Enhancer – Bremelanotide
Bremelanotide is a compound that is currently under investigation for its potential uses in managing reperfusion injury, female sexual dysfunction or hemorrhagic shock. The chemical may also see success in managing modulate inflammation or limiting the effects of ischemia.
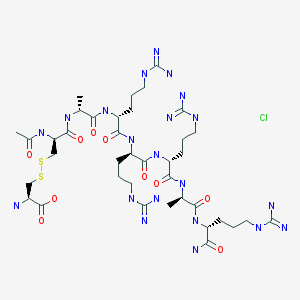
N-Acetyl-L-norleucyl-L-alpha-aspartyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-lysine (2-7)-lactam
Bremelanotide, PT 141, CAS NO.: 189691-06-3
Bremelanotide  i/ˌbrɛmɨˈlænətaɪd/ (formerly PT-141) is a compound under drug development by Palatin Technologies as a treatment for female sexual dysfunction, hemorrhagic shock and reperfusion injury. It functions by activating the melanocortin receptors MC1R and MC4R, to modulate inflammation and limiting ischemia.[2] It was originally tested for intranasal administration in treating female sexual dysfunction but this application was temporarily discontinued in 2008 after concerns were raised over adverse side effects of increased blood pressure. As of December 2014, Palatin is conducting a human Phase 3 study[3] using a subcutaneous drug delivery system that appears to have little effect on blood pressure.
i/ˌbrɛmɨˈlænətaɪd/ (formerly PT-141) is a compound under drug development by Palatin Technologies as a treatment for female sexual dysfunction, hemorrhagic shock and reperfusion injury. It functions by activating the melanocortin receptors MC1R and MC4R, to modulate inflammation and limiting ischemia.[2] It was originally tested for intranasal administration in treating female sexual dysfunction but this application was temporarily discontinued in 2008 after concerns were raised over adverse side effects of increased blood pressure. As of December 2014, Palatin is conducting a human Phase 3 study[3] using a subcutaneous drug delivery system that appears to have little effect on blood pressure.
Palatin, in collaboration with European licensee Gedeon Richter, is developing an sc formulation of the synthetic peptide bremelanotide (PT-141; BMT), a melanocortin MCR-4 agonist and a synthetically modified analog of PT-14, also analogous to alpha-melanocyte-stimulating hormone (alpha-MSH), for the potential treatment of female sexual dysfunction (FSD) including hypoactive sexual desire disorder (HSDD)

The Bremelanotide or PT-141 is a mean that explains the revolution caused by the medical world in a silent but attractive manner in the human health related study. Bremelanotide is the latest arrival from the company called Palatin Technologies which forms the basic treatment for the hemorrhagic shock and reperfusion injury.( In short about the company, the Palatin Technologies is the owner of this research and is located in New Jersey. Hence this medicine is a Jersey based Product. And regarding the product under research, is waiting for the approval from the Food and Drug Association. Once this is done, the company has targeted to reach those customers, whom the Viagra has approached. This has the effect of helping the male patients suffering with an erectile dysfunction syndrome. Also if it gets the approval as a treatment measure for the female sexual dysfunction, then this medicine is expected to bring a relief to the post-menopausal and also supports or provides their sexual happiness and also they are checking regarding thehyposexual desire disorder. This is expected to be a blockbuster, if released. So this medicine is waiting for a confirmation as well as an approval.
In February 2015, a randomized, double-blind, placebo-controlled, open-label extension, phase III trial (NCT02338960; BMT-302, Reconnect Study) was initiated in the US in premenopausal women (expected n = 550) with hypoactive sexual desire disorder to evaluate the efficacy and safety of bremelanotide. At that time, the trial was expected to complete in July 2017
Study – Potential Use Erectile Dysfunction
One study has explored the potential use of bremelanotide as a replacement for natural peptide melanocyte stimulating hormones for the sake of treating erectile dysfunction.
- The goal of this study was to determine if the effects of bremelanotide stimulating sexual desire that was shown in male rats could be replicated in the brains of female rats. To do this, hormone primed female rats in a control group and a test group that were treated with bremelanotide and known to have consummatory sexual disorders was introduced to a group of male rats and the reactions were measured.
- Heart racing, hops and darts, pacing and customary sexual behaviors were assessed while the brain was stimulated. The stimulation of specific molecular markers within the brain was examined to determine arousal in the female subjects.
- Results indicated that the females saw an increase in sexual behavior when bremelanotide was applied to the limbic and hypothalamic regions of their brains. It is suggested that this was because the chemical that stimulated the mPOA terminals, leading to activated dopamine in the brain.
Additional study is necessary to determine the extent of the effects bremelanotide has on the brain and natural stimulating chemicals.

Bremelanotide and Ongoing Research

This is an advanced research involved even now. This functions by activating the Melanocortin, which is a group of peptide hormones which includes the adrenocorticotropic hormone and also the different forms of the melanocyte stimulating hormones. These melanocortins are produced or prepared from the proopiomelanocortin in the pituitary glands. The melanocortin releases or exert their effects by making a bind with the melanocortin and thereby activating it).The Bremelanotide functions by activating the melanocortin receptors and thereby makes a modulation in the inflammation. This is actually produced for making use in treating the sexual dysfunction. Due to certain reasons; the process of researching was kept under hold in recently, since it created some adverse side effects of increased blood pressure. In the chemistry of the preparation of the bremelanotide, the Peptide Melanaton II forms the basic compound. This compound is tested using a sunless tanning agent.
The actual information about the peptide melanaton has the effect of making sexual arousal and speed as well as sudden erections and some other side effects. However, there are several other measures taken to test the property of the same under several other health situations to make a detailed study about the chemical compound structure to make a change in the combination of the chemical structure. This medicine has made a revolution in the field of science of the human structure. When made a deep verification of the compound structure of the chemical study showed the following information. The structural design has an appearance of white colored powder like material, which has an accurate purity of nearly 98%. The actual molecular weight of the compound formed is around 1025.2. This compound has the collective share of Amino acids in the composition, peptide and acetate contents also.
The study of the compound structure PT-141 has an enhanced support of making a recombination that produces a different profile of the same medicine but in a different standard with different properties that may support the human requirement.
Bremelanotide PT-141 is known for its aphrodisiac properties
Development
![]()
Bremelanotide was developed from the peptide hormone Melanotan II which underwent testing as a sunless tanningagent. In initial testing, Melanotan II did induce tanning but additionally caused sexual arousal and spontaneous erections as unexpected side effects in nine out of the ten original male volunteer test subjects.[4]
In studies, bremelanotide was shown to induce lordosis in an animal model[5] and was also effective in treating sexual dysfunction in both men (erectile dysfunction or impotence) and women (sexual arousal disorder). Unlike Viagra and other related medications, it does not act upon the vascular system, but directly increases sexual desire via the nervous system.[6]
A Phase III clinical trial was scheduled to begin in the first half of 2007, but was delayed until August 2007. On August 30, Palatin announced that the U.S. Food and Drug Administration had expressed serious concerns regarding therisk/benefit ratio of bremelanotide with regards to the side effect of increased blood pressure. The FDA stated that it would consider alternate uses for bremelanotide, including as a treatment for individuals who do not respond to more established ED treatments. However, On May 13, 2008, Palatin Technologies announced it had “discontinued development of Bremelanotide for the treatment of male and female sexual dysfunction” while concurrently announcing plans to develop it as a treatment for hemorrhagic shock instead.[7] The company additionally announced intentions to focus its attention on another compound, PL-6983, that causes lower blood pressure in animal models.[8]Palatin has since re-initiated Bremelanotide studies for ED and FSD using a subcutaneous delivery method. On August 12, 2009, the company announced that in a double-blind study of 54 volunteers bremelanotide failed to evoke the hypertensive side effects seen with the nasal delivery system used in prior studies, concluding that “variability of uptake” inherent in intranasal administration of the drug resulted in “increases in blood pressure and gastrointestinal events…primarily related to high plasma levels in [only] a subset of patients” and that subcutaneous administration of the drug circumvented the potential for this side effect.[8] Palatin has completed a human Phase 2B study utilizing subcutaneous administration and reported positive results.[9]
Structure
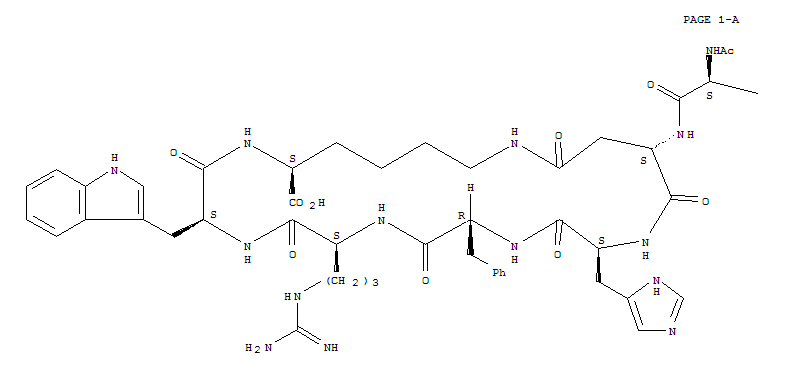
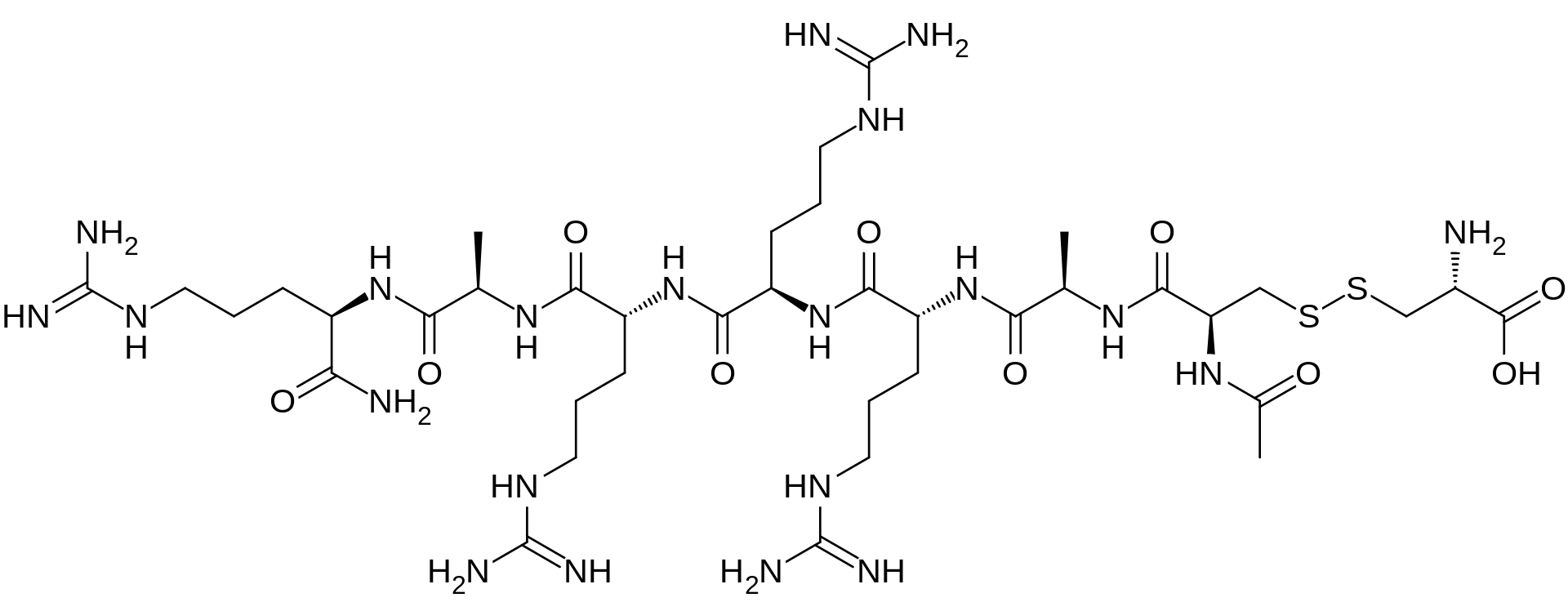
Bremelanotide is a cyclic hepta-peptide lactam analog of alpha-melanocyte-stimulating hormone (alpha-MSH) that activates the melanocortin receptors MC3-R and MC4-R in thecentral nervous system. It has the amino acid sequence Ac-Nle-cyclo[Asp-His-D-Phe-Arg-Trp-Lys]-OH or cyclo-[Nle4, Asp5, D-Phe7, Lys10]alpha-MSH-(4-10). It is a metabolite of Melanotan II that lacks the C-terminal amide function.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3S,6S,9R,12S,15S,23S)-15-[(N-acetyl-L-norleucyl)amino]-9-benzyl-6-{3-[(diaminomethylidene)amino]propyl}-12-(1H-imidazol-5-ylmethyl)-3-(1H-indol-3-ylmethyl)-2,5,8,11,14,17-hexaoxo-1,4,7,10,13,18-hexaa zacyclotricosane-23-carboxylic acid | |
| Clinical data | |
| Legal status |
|
| Pharmacokinetic data | |
| Half-life | 120 minutes[1] |
| Identifiers | |
| CAS number | 189691-06-3  |
| ATC code | None |
| PubChem | CID 9941379 |
| ChemSpider | 8116997 |
| UNII | 6Y24O4F92S |
| KEGG | D06569  |
| ChEMBL | CHEMBL2070241 |
| Chemical data | |
| Formula | C50H68N14O10 |
| Molecular mass | 1025.2 g/mol |
Sexual dysfunction, including both penile erectile dysfunction or impotence and female sexual dysfunction, are common medical problems. Significant effort has been devoted over the last twenty or more years to develop methods, devices and compounds for treatment of sexual dysfunction. While more effort has been undertaken for treatment of penile erectile dysfunction, female sexual dysfunction is also an area to which significant research and effort has been devoted.
At present, one commonly used orally administered drug for treatment of sexual dysfunction in the male is Viagra®, a brand of sildenafil, which is a phosphodiesterase 5 inhibitor, increasing the persistence of cyclic guanosine monophosphate and thereby enhancing erectile response. There are several other medical treatment alternatives currently available depending on the nature and cause of the impotence problem. Some men have abnormally low levels of the male hormone testosterone, and treatment with testosterone injections or pills may be beneficial. However, comparatively few impotent men have low testosterone levels. For many forms of erectile dysfunction, treatment may be undertaken with drugs injected directly into the penis, including drugs such as papaverin, prostaglandin E1, phenoxybenzamine or phentolamine. These all work primarily by dilating the arterial blood vessels and decreasing the venous drainage. Urethral inserts, such as with suppositories containing prostaglandin, may also be employed. In addition, a variety of mechanical aids are employed, including constriction devices and penile implants.
A variety of treatments have also been explored for female sexual dysfunction, including use of sildenafil, although the Food and Drug Administration has not specifically approved such use. Testosterone propionate has also been employed to increase or augment female libido.
Melanocortin receptor-specific compounds have been explored for use of treatment of sexual dysfunction. In one report, a cyclic α-melanocyte-stimulating hormone (“α-MSH”) analog, called Melanotan-II, was evaluated for erectogenic properties for treatment of men with psychogenic erectile dysfunction. Wessells H. et al., J Urology 160:389-393 (1998); see also U.S. Pat. No. 5,576,290, issued Nov. 19, 1996 to M. E. Hadley, entitled Compositions and Methods for the Diagnosis and Treatment of Psychogenic Erectile Dysfunction and U.S. Pat. No. 6,051,555, issued Apr. 18, 2000, also to M. E. Hadley, entitled Stimulating Sexual Response in Females. The peptides used in U.S. Pat. Nos. 5,576,290 and 6,051,555 are also described in U.S. Pat. No. 5,674,839, issued Oct. 7, 1997, to V. J. Hruby, M. E. Hadley and F. Al-Obeidi, entitled Cyclic Analogs of Alpha-MSH Fragments, and in U.S. Pat. No. 5,714,576, issued Feb. 3, 1998, to V. J. Hruby, M. E. Hadley and F. Al-Obeidi, entitled Linear Analogs of Alpha-MSH Fragments. Melanotan-II is a peptide of the following formula:

Additional related peptides are disclosed in U.S. Pat. Nos. 5,576,290, 5,674,839, 5,714,576 and 6,051,555. These peptides are described as being useful for both the diagnosis and treatment of psychogenic sexual dysfunction in males and females. These peptides are related to the structure of melanocortins.
In use of Melanotan-II, significant erectile responses were observed, with 8 of 10 treated men developing clinically apparent erections, and with a mean duration of tip rigidity greater than 80% for 38 minutes with Melanotan-II compared to 3.0 minutes with a placebo (p=0.0045). The drug was administered by subcutaneous abdominal wall injection, at doses ranging from 0.025 to 0.157 mg/kg body weight. Transient side effects were observed, including nausea, stretching and yawning, and decreased appetite.
The minimum peptide fragment of native α-MSH needed for erectile response is the central tetrapeptide sequence, His6-Phe7-Arg8-Trp9 (SEQ ID NO:1). In general, all melanocortin peptides share the same active core sequence, His-Phe-Arg-Trp (SEQ ID NO:1), including melanotropin neuropeptides and adrenocorticotropin. Five distinct melanocortin receptor subtypes have been identified, called MC1-R through MC5-R, and of these MC3-R and MC4-R are believed to be expressed in the human brain. MC3-R has the highest expression in the arcuate nucleus of the hypothalamus, while MC4-R is more widely expressed in the thalamus, hypothalamus and hippocampus. A central nervous system mechanism for melanocortins in the induction of penile erection has been suggested by experiments demonstrating penile erection resulting from central intracerebroventricular administration of melanocortins in rats. While the mechanism of His-Phe-Arg-Trp (SEQ ID NO:1) induction of erectile response has not been fully elucidated, it has been hypothesized that it involves the central nervous system, and probably binding to MC3-R and/or MC4-R.
Other peptides and constructs have been proposed which are ligands that alter or regulate the activity of one or more melanocortin receptors. For example, International Patent Application No. PCT/US99/09216, entitled Isoquinoline Compound Melanocortin Receptor Ligands and Methods of Using Same, discloses two compounds that induce penile erections in rats. However, these compounds were administered by injection at doses of 1.8 mg/kg and 3.6 mg/kg, respectively, and at least one compound resulted in observable side effects, including yawning and stretching. Other melanocortin receptor-specific compounds with claimed application for treatment of sexual dysfunction are disclosed in International Patent Application No. PCT/US99/13252, entitled Spiropiperidine Derivatives as Melanocortin Receptor Agonists.
Both cyclic and linear α-MSH peptides have been studied; however, the peptides heretofore evaluated have had an amide or —NH2 group at the carboxyl terminus. See, for example, Wessells H. et al., J Urology, cited above; Haskell-Luevano C. et al., J Med Chem 40:2133-39 (1997); Schiöth H. B. et al., Brit J Pharmacol 124:75-82 (1998); Schiöth H. B. et al., Eur J Pharmacol 349:359-66 (1998); Hadley M. E. et al., Pigment Cell Res 9:213-34 (1996); Bednarek M. A. et al., Peptides20:401-09 (1999); U.S. Pat. Nos. 6,054,556, 6,051,555 and 5,576,290; and, International Patent Applications PCT/US99/04111 and PCT/US98/03298. While significant research has been conducted in an effort to determine the optimal structure of α-MSH peptides, including a variety of structure-function, agonist-antagonist, molecular modeling and pharmacophore studies, such studies have relied upon peptides with an art conventional —NH2 group at the carboxyl terminus. Further, it has long been believed that biologically active neuropeptides, including α-MSH peptides, are amidated, with an —NH2 group at the carboxyl terminus, and that such amidation is required both for biological activity and stability. See, for example, Metabolism of Brain Peptides, Ed. G. O’Cuinn, CRC Press, New York, 1995, pp. 1-9 and 99-101.
…………………………………………….
Bioorganic and Medicinal Chemistry Letters, 2005 , vol. 15, 4 pg. 1065 – 1068
http://www.sciencedirect.com/science/article/pii/S0960894X04014842

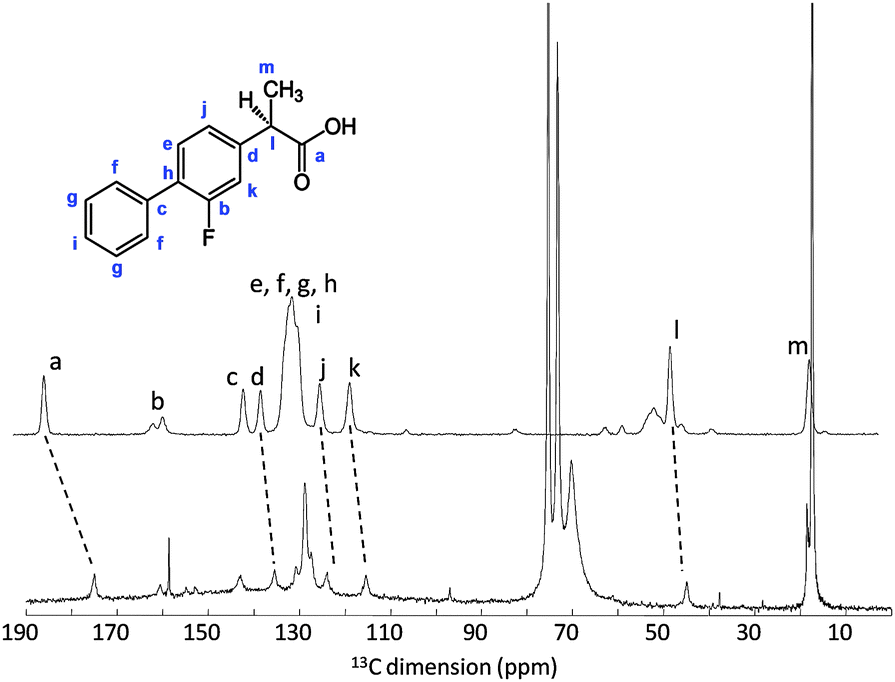
NMR structural analysis on compound 3.

NMR structural analysis of compound 1.
……………………………………………….
In a preferred embodiment, the invention provides the peptide
Ac-Nle-cyclo(-Asp-His-D-Phe-Arg-Trp-Lys)-OH Compound 1
The peptide of Compound 1 has a formula of C50H68N14O10, and a net molecular weight of 1025.18. This peptide may be synthesized by solid-phase means and purified to greater than 96% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of Compound 1 is:

In general, the peptide compounds of this invention may be synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of well-known procedures utilizing a variety of resins and reagents may be used to prepare the compounds of this invention.
The peptides of this invention may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacetic, maleic, succinic or methanesulfonic. The acetate salt form is especially useful. Where the compounds of this invention include an acidic moiety, suitable pharmaceutically acceptable salts may include alkali metal salts, such as sodium or potassium salts, or alkaline earth metal salts, such as calcium or magnesium salts.
The invention provides a pharmaceutical composition that includes a peptide of this invention and a pharmaceutically acceptable carrier. The carrier may be a liquid formulation, and is preferably a buffered, isotonic, aqueous solution. Pharmaceutically acceptable carriers also include excipients, such as diluents, carriers and the like, and additives, such as stabilizing agents, preservatives, solubilizing agents, buffers and the like, as hereafter described.
EXAMPLE 1
Peptide Synthesis
The peptide Ac-Nle-cyclo(-Asp-His-D-Phe-Arg-Trp-Lys)-OH was synthesized by standard solid phase peptide synthesis methods, and is a cyclic heptapeptide melanocortin peptide analog with a free acid at the carboxyl terminus and an acetylated amino group at the amino terminus, with the structure:

The peptide has a net molecular weight of 1025.18, and is supplied in an acetate salt form. The peptide is a white, odorless amorphous hygroscopic powder, soluble in 0.9% saline, composed of C50H68N14O10. For synthesis, an Fmoc-Lys(R3)-p-alkoxybenzyl alcohol resin was transferred to a solid phase peptide synthesizer reactor with a mechanical stirrer. The R3group, such as 1-(1′-adamantyl)-1-methyl-ethoxycarbonyl (Adpoc), allyloxycarbonyl (Aloc) or 4-methyltrityl (Mtt), was removed and the next Fmoc-protected amino acid (Fmoc-Trp(Boc)-OH) was added to the resin through standard coupling procedures. The Fmoc protective group was removed and the remaining amino acids added individually in the correct sequence, by repeating coupling and deprotection procedures until the amino acid sequence was completed. After completion of coupling with the last Fmoc-amino acid derivative, Fmoc-Nle-OH, and cleavage of the Fmoc protective group, the exposed terminal amino group was acetylated with acetic anhydride and pyridine in N,N-dimethylformamide (DMF). The peptide-resin was dried and the Lys and Asp protective groups cleaved. The Lys and Asp deprotected peptide resin was suspended in a suitable solvent, such as DMF, dichloromethane (DCM) or 1-methyl-2-pyrrolidone (NMP), a suitable cyclic coupling reagent, such as 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU), 2-(7-aza-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TATU), 2-(2-oxo-1(2H)-pyridyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) or N,N′-dicyclohexylcarbodiimide/1-hydroxybenzotriazole (DCCl/HOBt) was added, and coupling initiated by use of a suitable base, such as N,N-diispropylethylamine (DIPEA), sym-collidine or N-methylmorpholine (NMM). After cyclization, the peptide-resin was washed and the peptide cleaved from the resin and any remaining protective groups using trifluoroacetic acid (TFA) in the presence of water and 1,2-ethanedithiol (EDT). The final product was precipitated by adding cold ether and collected by filtration. Final purification was by reversed phase HPLC using a C18 column. The purified peptide was converted to acetate salt by passage through an ion-exchange column.
…………………………………………..
WO2014071339
Compounds of the Invention.
in a preferred embodiment of the present invention, fie rneianocortin receptor agonist is;
Ac-Nie”Cyc/o{-Asp-His–D–Phe-Arg–Trp»Lys)–OH (bremeianotide)
The peptide of bremeianotide has a formula of CsaHesN< C½, and a net mofecufar weight of 1025.18, This peptide may be synthesized by conventional means, including either solid-phase or Squid-phase techniques, and purified to greater than 99% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of bremeianotide is:

in one embodiment of the invention, bremeianotide is synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of ‘well-known procedures utilizing a variety of resins and reagents may be used to prepare bremeianotide.
Bremeianotide may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacefie, maieic, citric, tartaric, oxalic, succinic or methanesu!fonic acid. The acetate salt form is especially useful.
in a preferred embodiment, bremelanotide is an acetate salt form, and is formulated in a buffered aqueous solution including giycerin, and prepackaged in a syringe and auto-injector device. In alternative embodiments, bremelanotide is any pharmaceutically acceptable salt form, and is formulated in any pharmaceutically acceptable aqueous solution, the aqueous solution optionally including one or more salts, such as sodium chloride, one or more acids, such as citric acid, and one or more additional ingredients, including cellulose or derivatives thereof, saccharides o
polysaccharides such as dextrose, and any of a wide variety of surfactants, chelating agents and preservatives.
………………………………………….
In yet another embodiment of the present invention, the melanocortin receptor agonist is:
Ac-Nle-cyclo(-Asp-His-D-Phe-Arg-Trp-Lys)-OH PT-141
The peptide of PT-141 has a formula of C50H68N14O10, and a net molecular weight of 1025.18. This peptide may be synthesized by conventional means, including either solid-phase or liquid-phase techniques, and purified to greater than 99% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of PT-141 is:

In one embodiment of the invention, PT-141 is synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of well-known procedures utilizing a variety of resins and reagents may be used to prepare PT-141.
PT-141 may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacetic, maleic, citric, tartaric, oxalic, succinic or methanesulfonic acid. The acetate salt form is especially useful. Where the compounds of this invention include an acidic moiety, suitable pharmaceutically acceptable salts may include alkali metal salts, such as sodium or potassium salts, or alkaline earth metal salts, such as calcium or magnesium salts.
In a preferred embodiment, PT-141 is an acetate salt form, and is formulated in a buffered aqueous solution including glycerin, prepackaged in a metered unit dose intranasal delivery device. In alternative embodiments, PT-141 is any pharmaceutically acceptable salt form, and is formulated in any pharmaceutically acceptable aqueous solution, the aqueous solution optionally including one or more salts, such as sodium chloride, one or more acids, such as citric acid, and one or more additional ingredients, including cellulose or derivatives thereof, saccharides or polysaccharides such as dextrose, and any of a wide variety of surfactants, chelating agents and preservatives. In one preferred embodiment, PT-141 is administered to patients in volumes of 100 μL, with the quantity of PT-141 delivered determined by the concentration thereof. As described hereafter, in one preferred embodiment a metered unit dose contains 7.5 mg of PT-141.
While certain embodiments of the present invention are described primarily in the context of PT-141, it is to be understood that other melanocortin receptor agonists may be employed. For example, the metallopeptide melanocortin receptor agonists disclosed in WO 02/064091, filed on Feb. 13, 2001, and U.S. Ser. No. 10/640,755, filed on Aug. 13, 2003, both entitled Melanocortin Metallopeptides for Treatment of Sexual Dysfunction; and WO 01/13112, filed on Jun. 14, 2000, entitled Melanocortin Metallopeptide Constructs, Combinatorial Libraries and Applications, may be employed. In addition, the peptidomimetic melanocortin receptor agonists disclosed in U.S. Ser. No. 10/776,419, filed on Feb. 10, 2004, entitled Peptidomimetics of Biologically Active Metallopeptides; the pyrrolidine melanocortin receptor agonists disclosed in U.S. Ser. No. 10/766,657, filed on Feb. 10, 2004, entitled Pyrrolidine Melanocortin-Specific Compounds; and the bicyclic melanocortin receptor agonists disclosed in PCT/US04/01505, filed on Jan. 20, 2004, entitled Bicyclic Melanocortin-Specific Compounds, may also be employed. Also particular preferred are the piperazine melanocortin agonists disclosed in PCT/US04/01462, filed on Jan. 20, 2004 and U.S. Ser. No. 10/762,079, filed on Jan. 20, 2004, both entitled piperazine Melanocortin-Specific Compounds; the melanocortin agonists disclosed in WO 03/006620, filed on Jul. 11, 2002, entitled Linear and Cyclic Melanocortin Receptor-Specific Peptides; WO 04/005324, filed on Jul. 9, 2003, entitled Peptide Compositions for Treatment of Sexual Dysfunction; PCT/US00/18217, filed on Jun. 29, 2000 and U.S. Ser. No. 10/040,547, filed on Jan. 4, 2002, entitled Compositions and Methods for Treatment of Sexual Dysfunction; and U.S. Ser. No. 10/638,071, filed on Aug. 8, 2003, entitled Cyclic Peptide Compositions and Methods for Treatment of Sexual Dysfunction. The entire disclosure of each of the foregoing are incorporated here by reference. It is to be understood that the foregoing listing of patent applications disclosing melanocortin receptor agonists is intended to only be exemplary, and that other melanocortin receptor agonists, whether heretofore known or hereafter developed, may similarly be used in the practice of this invention.
…………………….
NMR prediction


- King SH, Mayorov AV, Balse-Srinivasan P, Hruby VJ, Vanderah TW, Wessells H (2007).“Melanocortin receptors, melanotropic peptides and penile erection”. Current Topics in Medicinal Chemistry 7 (11): 1098–1106. doi:10.2174/1568026610707011111.PMC 2694735. PMID 17584130.
- Bremelanotide for Organ Protection and Related Indications, Palatin Technologies fact sheet. Retrieved on 2009-01-18.
- “Palatin Announces Start of Bremelanotide Phase 3 Program For Female Sexual Dysfunction”. PR Newswire. Retrieved 2015-02-17.
- “Tanning drug may find new life as Viagra alternative”. CNN. 1999. Retrieved2007-09-16.
- Pfaus JG, Shadiack A, Van Soest T, Tse M, Molinoff P (July 2004). “Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist”. Proc. Natl. Acad. Sci. U.S.A. 101 (27): 10201–4. doi:10.1073/pnas.0400491101. PMC 454387.PMID 15226502.
- Vicki Mabrey (2006). “ABC News “The Business of Desire – Love Potion””. ABC News. Retrieved 2009-01-24.
- “Palatin Technologies announces new strategic objectives and reports third quarter 2008 financial results”. Palatin Technologies press release. 2008. Retrieved 2008-08-21.
- “Palatin Technologies Announces New Strategic Objectives”. Retrieved 2008-05-13.
- http://www.palatin.com/news/news.asp?ud=306
External links
- Palatin Technologies The company that has developed bremelanotide.
- US 6,794,489 bremelanotide (PT-141) patent (Appl. No.:040547)
- US 6,579,968 bremelanotide (PT-141) patent (Appl. No.:066501)
- Bremelanotide Edguider Forum
| PALATIN TECHNOLOGIES, INC.: ‘Bremelanotide in Premenopausal Women With Female Sexual Arousal Disorder and/or Hypoactive Sexual Desire Disorder‘ CLINICALTRIALS.GOV (NCT01382719, [Online] 20 March 2012, page 1 Retrieved from the Internet: <URL:http://clinicaltrials.gov/archive/NCT0 1382719/ 2012-03 20> [retrieved on 2014-02-10] | ||
| 2 | * | PALATIN TECHNOLOGIES, INC.: ‘Reports Positive Bremelanotide Study; Improved Safety Profile with Subcutaneous Administration‘ PR NEWSWIRE., [Online] 12 August 2009, Retrieved from the Internet: <URL:http://www.thefreelibrary.com/Palatin +Technolo9ies,+Inc.+Reports+Positive+Bremel anotide+Study%38…-a020561 3302> [retrieved on 2014-02-10] |
| 3 | * | SAFARINEJAD, MR.: ‘Evaluation of the Safety and Efficacy of Bremelanotide, a Melanocortin Receptor Agonist, in Female Subjects with Arousal Disorder: A Double-Blind Placebo-Controlled, Fixed Dose, Randomized Study”.‘ INTERNATIONAL SOCIETY FOR SEXUAL MEDICINE. vol. 5, 2008, pages 887 – 897 |
| US8455617 | Jun 7, 2010 | Jun 4, 2013 | Astrazeneca Ab | Melanocortin receptor-specific peptides |
| US8455618 | Oct 26, 2011 | Jun 4, 2013 | Astrazeneca Ab | Melanocortin receptor-specific peptides |
| US8487073 | Nov 23, 2010 | Jul 16, 2013 | Palatin Technologies, Inc. | Melanocortin receptor-specific peptides for treatment of sexual dysfunction |
| US8729224 | Jun 5, 2013 | May 20, 2014 | Palatin Technologies, Inc. | Melanocortin receptor-specific peptides for treatment of female sexual dysfunction |
| EP2266567A1 | May 26, 2009 | Dec 29, 2010 | Æterna Zentaris GmbH | Use of cetrorelix in combination with PDE V inhibitors for the treatment of sex hormone dependent disorders |
| EP2266568A1 | May 26, 2009 | Dec 29, 2010 | Æterna Zentaris GmbH | Use of LHRH antagonists in combination with PDE V inhibitors for the treatment of sex hormone dependent disorders |
| WO2013067309A1 | Nov 2, 2012 | May 10, 2013 | Xion Pharmaceutical Corporation | Methods and compositions for oral administration of melanocortin receptor agonist compounds |
| WO2014071339A2 * | Nov 5, 2013 | May 8, 2014 | Palatin Technologies, Inc. | Uses of bremelanotide in therapy for female sexual dysfunction |
| WO2009151714A2 * | Mar 24, 2009 | Dec 17, 2009 | Palatin Technologies, Inc. | Therapeutic for treatment of circulatory shock, ischemia, inflammatory disease and related conditions |
| US6794489 * | Jan 4, 2002 | Sep 21, 2004 | Palatin Technologies, Inc. | Peptide sequence ac-nle-cyclo(-asp-his-d-phe-arg-trp-lys)-oh derived from a melanocyte-stimulating hormone (? alpha -msh?) analog, called melanotan-ii |
| US20050222014 * | May 26, 2005 | Oct 6, 2005 | Palatin Technologies, Inc. | Administering phosphodiestarase inhibitors and melanocortin receptor antagonist: synergistic mixture |
| US20110065652 * | Nov 23, 2010 | Mar 17, 2011 | Palatin Technologies, Inc. | Melanocortin Receptor-Specific Peptides for Treatment of Sexual Dysfunction |
Filed under: Phase3 drugs, Premture ejaculation, sex arousal Tagged: Bremelanotide, Female Libido Enhancer, Inc., Palatin Technologies, PHASE 3, PT 141, sex arousal








































































 .
.
 .
.

























 .
. .
.
























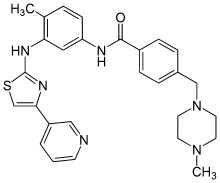
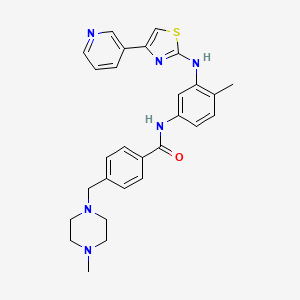
![methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide NMR spectra analysis, Chemical CAS NO. 1048007-93-7 NMR spectral analysis, methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-15/001/603/1603135_1h.png)
![methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide NMR spectra analysis, Chemical CAS NO. 1048007-93-7 NMR spectral analysis, methanesulfonic acid,4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-yl-1,3-thiazol-2-yl)amino]phenyl]benzamide C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2015-01-15/001/603/1603135_13c.png)
































































 LIONEL MY SON
LIONEL MY SON















 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..



 amcrasto@gmail.com
amcrasto@gmail.com LIONEL MY SON
LIONEL MY SON


































































-9,10-di((2H3)methoxy)-3-(2-methylpropyl)-1,3,4,6,7,11b-hexahydro-2H-benzo(a)quinolizin-2-one")




















































 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..

 amcrasto@gmail.com
amcrasto@gmail.com



















%20structure.gif)























































































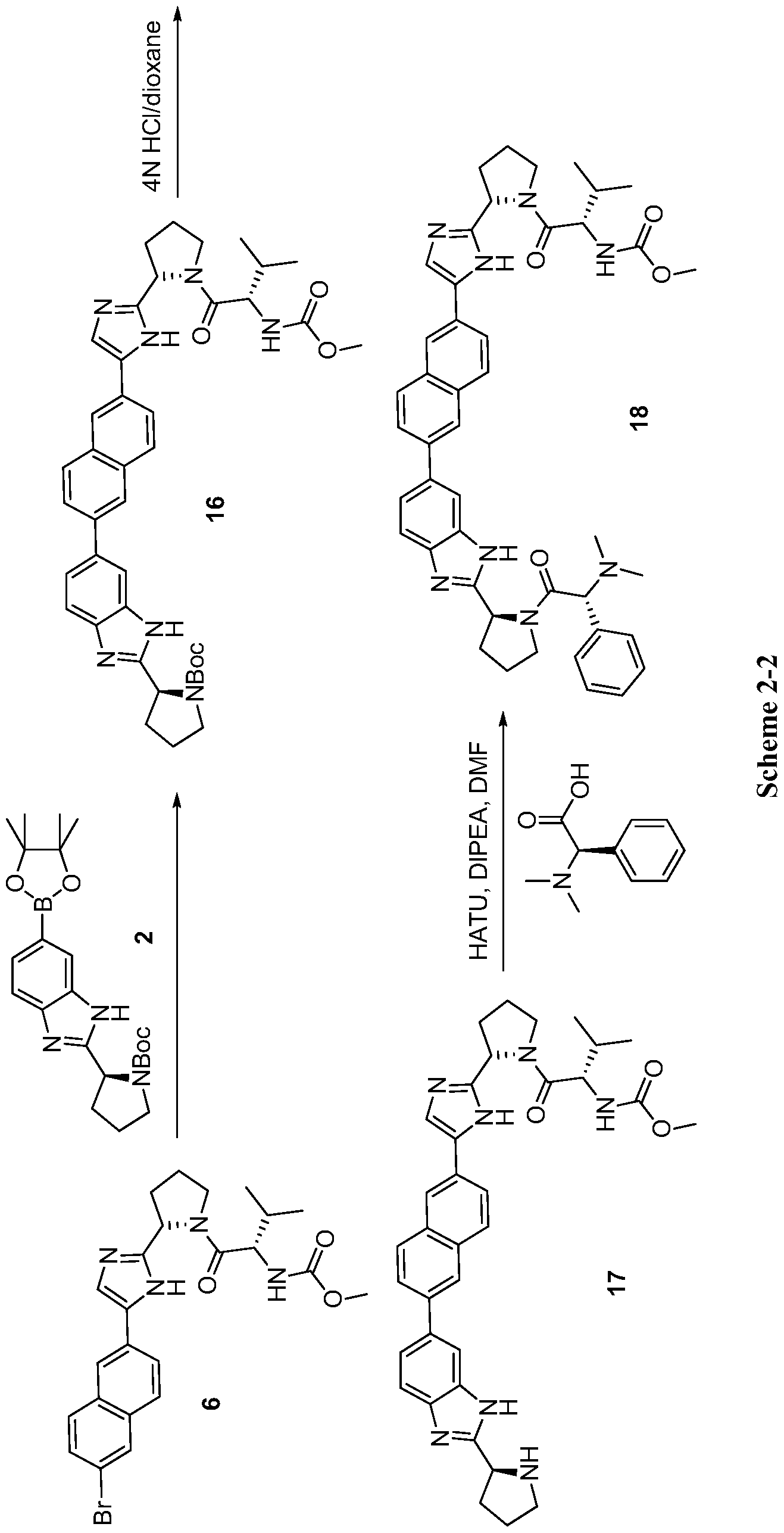
 scheme 2-3
scheme 2-3







































Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.